About this method
E-SNPs&GO is a fast and accurate method that, given an input protein sequence and a single residue variation, can predict whether the variation is related to diseases or not. The prediction relies on an input encoding completely based on protein language models and embedding techniques, specifically devised to encode protein sequences and GO functional annotations. We trained our model on a newly generated dataset of 101,146 human protein single residue variants derived from public resources. When tested on a blind set comprising 10,266 variants, 4,083 of which are pathogenic and 6,183 benign, our method reaches an MCC score of 0.72.
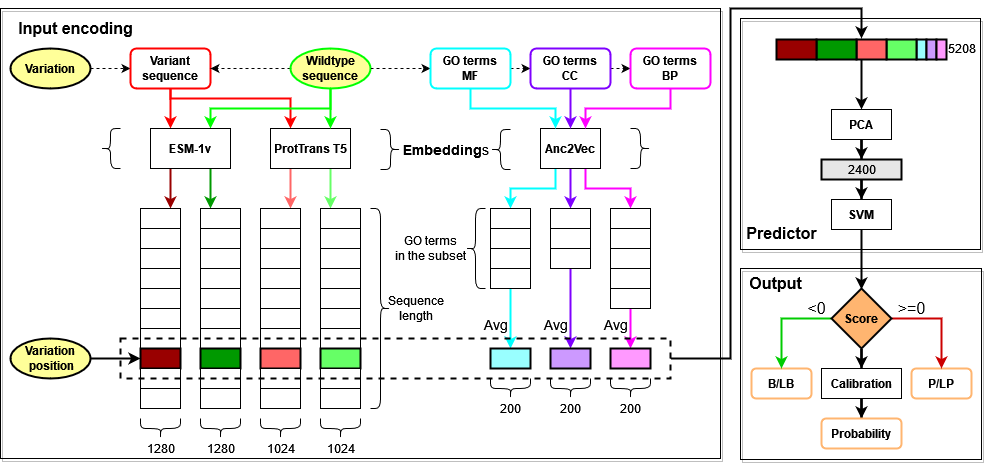
Given a protein sequence and a variation occurring at a specific position, both the wildtype and the variant sequences are embedded using two Protein Language Models, ESM-1v and ProtTrans T5, generating 4 matrices from which we extract only the vectors corresponding to the mutated position. We also extract GO functional annotations from the wildtype sequence that we embed with a third model called Anc2Vec, averaging together all annotations belonging to the same sub-ontology (Molecular Function, Cellular Component, Biological Process). Each variation is finally encoded with a vector comprising 2*1280 + 2*1024 + 3*200 = 5208 features, to which we apply Principal Component Analysis for dimensionality reduction before feeding it to a Support Vector Machine that performs the binary classification task. The output score is also used for generating a calibrated Pathogenicity probability and a Reliability Index.